

Part 1: Foundations in the Science behind Sturge-Weber Syndrome

Hi, I’m Aron Thomassen, an undergraduate medicine offer-holder at King’s College London with a strong interest in paediatric neurology and translational neuroscience. This post was developed from my Extended Project Qualification (EPQ) and additional independent research. While I am not a qualified medical professional, I have made every effort to ensure this summary is accurate, well-referenced, and accessible to a wide audience. It is intended to inform and raise awareness, not to replace medical advice. I am always open to any feedback as to how I can improve, feel free to email me at aron.thomassen@icloud.com.
TL;DR
Sturge-Weber Syndrome is caused by a post-zygotic mutation in the GNAQ Gene, disrupting normal vascular regression in embryonic development. This results in neurocutaneous symptoms such as seizures, port-wine stains, and glaucoma. While there is no cure, multidisciplinary treatment is key to improving outcomes.
Introduction
Sturge-Weber Syndrome (SWS), otherwise known as encephalotrigeminal angiomatosis, is a rare congenital neurocutaneous disorder, most visibly recognised by a port-wine stain. However, the true complexity of SWS does not lie in just the skin, but rather in its complex neurological sequelae and unique clinical challenges. In this post, we explore how a single somatic mutation of the GNAQ gene disrupts the neurovascular development in the embryonic brain, leading to profound effects on patients’ neurological, cognitive and visual development.
i. Understanding the Basics
Sturge-Weber Syndrome is classified as a neurocutaneous disorder, a group of conditions that affect the nervous system ‘neuro’, and the skin ‘cutaneous’. SWS is a non-inheritable disease that arises from a somatic mosaic mutation of the GNAQ gene, meaning it happens post-fertilisation, and is only present in specific cells that have arisen from the original mutated cell, typically happening during early embryonic development.
The condition was named after William Allen Sturge and Frederick Parkes Weber, classically defined by the triad of:
A facial port-wine stain, usually in the V1 (ophthalmic nerve) branch of the trigeminal nerve.

By Madhero88 - Own work, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=6777419

Leptomeningeal Angiomas, which are vascular malformations where there are too many capillaries in the meninges of the brain.
Ocular Involvement, e.g glaucoma (frequent but not always involved).
ii. Embryological Origin
The origins of SWS lie in the early embryonic vascular development. At around week 6 of gestation, a vascular plexus forms in the neural tube, which is a network of capillaries interwoven with each other in order to supply oxygen and other nutrients to the cells to support the growth and development of the growing brain. Typically this plexus would regress by week 9 via cell signalling facilitated by the Gαq protein (the protein that the GNAQ gene creates).
However, in SWS, the somatic mutation of this GNAQ gene (typically the R183Q mutation) will change the structure of this protein, inhibiting its function, and therefore inhibiting the regression of the vascular plexus. As a result of this, the vascular tissue persists abnormally in regions of the body that derive from neural crest cells, which can include:
Facial Skin (presents as a port-wine stain)
Cerebral cortex (presents as a Leptomeningeal angioma)
The earlier that this mutation occurs during development, the more cells that it will affect, therefore leading to larger/more extensive clinical presentations. As a result of the mosaic nature of the mutation, the variability of this condition is remarkably large due to the multitude of factors that will affect the presentation.
iii. Clinical Features and Pathophysiology
Port-Wine Stain
A port-wine stain is a common presentation of SWS, caused by a capillary malformation of the dermis layer of skin, leading to the blood vessels being more dilated. It presents as a purple patch of the skin, often deepening in colour with age.
Leptomeningeal Angiomas and Cerebral Effects
Arguably the most damaging manifestation of SWS is the leptomeningeal angioma. Similar to the Port-Wine Stain, the leptomeningeal angioma is formed as a result of the mutated GNAQ gene leading to denser networks of capillaries, in this case in the meninges. This can initiate a cascade of pathological events, beginning with:
The leptomeningeal angioma impairs venous drainage from the cerebral cortex, particularly through the superficial cortical veins (located in the meninges) as they are often absent, underdeveloped or obstructed.
Consequently chronic venous hypertension and venous stasis occurs as there is reduced and sometimes stagnant blood flow.
The impaired drainage of blood elevates the pressure in the veins, disrupting the pressure gradients and causing the blood to flow slower in the arteries to the brain, causing chronic cerebral hypoperfusion.
This results in ischema as there is reduced delivery of oxygen and nutrients to brain cells, over time leading to progressive neuronal loss and well as progressive cortical atrophy.
The result of these events, this can lead to:
Seizures (which are often begin as focal and frequently drug-resistant).
Hemiparesis, which is weakness/ loss of sensation of one side of the body (often the opposite side of the body to where the leptomeningeal angioma is located).
Neurodevelopmental difficulties including cognitive delay, attention deficits and mood disturbances. Though these presentations significantly vary based on the extent and location of cortical involvement of SWS.
Epilepsy can play a huge role in the development of a child's brain. Many factors such as the age of onset, types of seizures as well as the frequency of seizures all play a role in the extent of any potential cognitive deficits (PMID: 29414553). In a study of children with unilateral SWS (only affects one side of the brain), researchers found a strong inverse relationship between age of seizure onset and cognitive performance across several domains, including Global IQ (GIQ), Verbal IQ (VIQ) and non-verbal IQ (PIQ). Specifically, the earlier a child experienced their first seizure, the greater the risk of cognitive deficits across all domains. These findings exemplify the importance of early detection and prevention of seizures, with the goal of maximising long-term developmental outcomes and quality of life.
iv. Diagnosis
Patients suspected of SWS will have a CT or MRI scan. This will now typically be done if the patient is seen to have a Port-Wine Stain in the V1 distribution to assess for associated involvement in the brain.
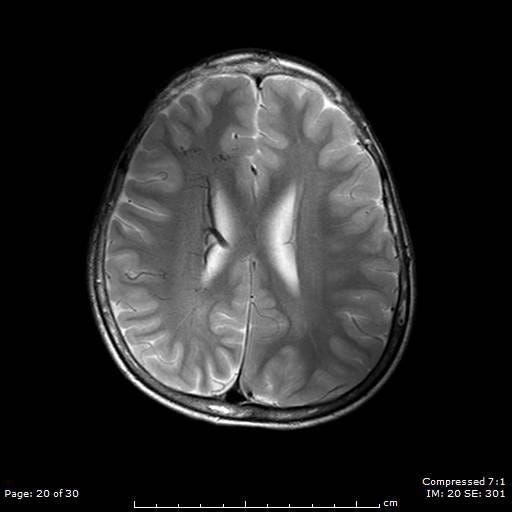
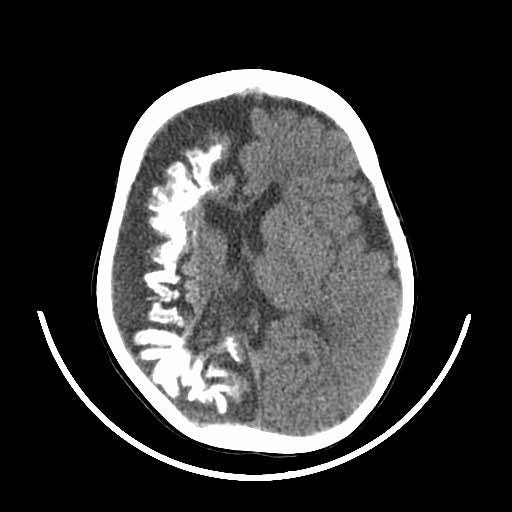
The image above shows an example of what an MRI (top) and a CT scan (bottom) would look like in a patient with SWS. In the CT scan on the left side, you can see this white mark. This is caused by a process known as calcification where deposits of calcium phosphate build up in the brain, creating ‘brain stones’ that can increase in size, and potentially induce further neurological issues. In addition, you are also able to observe the size difference from the left to the right of the brain. This is known as brain atrophy. Both calcification and extent of brain atrophy are key factors in determining the severity of the neurological conditions in SWS. Emerging research by Great Ormond Street Hospital proposed targeting these abnormally high inter-cranial calcium deposits as a potential therapeutic pathways to reduce progression of cortical damage.
v. Management
Sturge-Weber Syndrome does not have a ‘cure’, so it is of great importance that the symptoms of this condition are continuously managed to limit the long-term effects. As no two cases are the same, it requires nuanced multidisciplinary approach that involves:
Neurology & Neurosurgery to prescribe and monitor the affects of anti-epileptic drugs, and in some cases epilepsy surgery can be done to attempt to limit or stop seizures.
Dermatology to monitor the port-wine stain with the potential for Pulsed-dye laser to dampen the appearance of the birthmark.
Ophthalmology to monitor and attempt to halt the progression of any ocular manifestations.
Paediatrics: To give support to patients with neurodevelopmental delays.
And potentially many more, relating to the extent of the clinical manifestations, these can include speech and language therapy as well as occupational therapy.
vi. Conclusion
Sturge-Weber Syndrome stands as a profound example of how a single mutation during the development of a child can manifest across multiple systems, from the skin to the eyes and brain, producing a wide, and very variable array of clinical presentations. For clinicians, managing SWS means not only treating seizures or monitoring vision, but coordinating the care across an array of different specialities from neurology to ophthalmology to meet each patient’s unique needs.
Although a definitive cure remains not yet within reach, modern advances in imaging, surgical techniques and developmental care have significantly improved long-term outcomes. Early diagnosis, close surveillance, and family centred care still remain the most vital pillars for effective management of the disease.
As the understanding of the condition deepen in both the lab and clinic, the goal is not only to treat the visible signs of SWS, but also to support the patients in navigating the lifelong, complex journey of care this syndrome often entails.
References
Shirley, Matthew D., et al. “Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ.” The New England Journal of Medicine, vol. 368, no. 21, 23 May 2013, pp. 1971–1979, www.ncbi.nlm.nih.gov/pmc/articles/PMC3749068/, https://doi.org/10.1056/NEJMoa1213507. Accessed 26 May. 2025.
Comi, A.M. “Topical Review: Pathophysiology of Sturge-Weber Syndrome.” Journal of Child Neurology, 2003, www.semanticscholar.org/paper/Topical-Review%3A-Pathophysiology-of-Sturge-Weber-Comi/b9d19695e79ad656afe90d814b7228a5e42f3758, https://doi.org/10.1177/08830738030180080701.
Luat, Aimee F., et al. “Cognitive and Motor Outcomes in Children with Unilateral Sturge–Weber Syndrome: Effect of Age at Seizure Onset and Side of Brain Involvement.” Epilepsy & Behavior, vol. 80, Mar. 2018, pp. 202–207, www.ncbi.nlm.nih.gov/pmc/articles/PMC5845773/, https://doi.org/10.1016/j.yebeh.2018.01.012. Accessed 29 May. 2025.
“Sturge-Weber Syndrome: Video, Causes, & Meaning | Osmosis.” Osmosis, Elsevier, 2025, www.osmosis.org/learn/Sturge-Weber_syndrome. Accessed 29 May 2025.
‘Researchers design potential therapy to prevent brain deterioration in children with rare genetic conditions’ (2021) Crick. Available at: https://www.crick.ac.uk/news-and-reports/2023-10-04_researchers-design-potential-therapy-to-prevent-brain-deterioration-in-children-with-rare-genetic-conditions (Accessed: 27 May 2025).
Knöpfel, N. et al. (2023) ‘GNAQ/gna11 mosaicism is associated with abnormal serum calcium indices and microvascular Neurocalcification’, Journal of Investigative Dermatology [Preprint]. doi:10.1016/j.jid.2023.09.008.
Port-Wine Stains’ (2018) NHS, Great Ormond Street Hospital for Children. Available at: https://www.gosh.nhs.uk/conditions-and-treatments/conditions-we-treat/port-wine-stains/. (Accessed: 26 May 2025)
Male, R. and Eriksson, S.H. (2023) ‘The natural history of epilepsy and nonepileptic seizures in Sturge–Weber Syndrome: A retrospective case-note review’, Epilepsy and Behavior, 145, p. 109303. doi:10.1016/j.yebeh.2023.109303.
Tuxhorn, I.E.B. and Pannek, H.W. (2002) ‘Epilepsy surgery in bilateral Sturge-Weber Syndrome’, Pediatric Neurology, 26(5), pp. 394–397. doi:10.1016/s0887-8994(01)00414-3.
Harmon, K.A. et al. (2019) ‘Quality of life in children with Sturge-Weber Syndrome’, Pediatric Neurology, 101, pp. 26–32. doi:10.1016/j.pediatrneurol.2019.04.004.
The maxillary division of the trigeminal nerve (CNV2) TeachMeAnatomy. Available at: https://teachmeanatomy.info/head/nerves/maxillary-nerve/ (Accessed: 29 May 2025)
Bodensteiner, J.B. and Ewell Steve Roach (2010). Sturge-Weber Syndrome. 2nd ed. The Sturge-Weber Foundation.
Teh, D. (2023) Sturge-Weber Syndrome: Radiology reference article, Radiopaedia. Available at: https://radiopaedia.org/articles/sturge-weber-syndrome-1 (Accessed: 25 May 2025)
Loeb, J.A. et al. (2006) ‘Brain calcifications induce neurological dysfunction that can be reversed by a bone drug’, Journal of the Neurological Sciences, 243(1–2), pp. 77–81. doi:10.1016/j.jns.2005.11.033.